INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL
REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN
USE
ICH HARMONISED TRIPARTITE GUIDELINE
SPECIFICATIONS: TEST PROCEDURES AND ACCEPTANCE CRITERIA
FOR NEW DRUG SUBSTANCES AND NEW DRUG PRODUCTS:
CHEMICAL SUBSTANCES
Q6A
Current Step 4 version
dated 6 October 1999
This Guideline has been developed by the appropriate ICH Expert Working Group and
has been subject to consultation by the regulatory parties, in accordance with the ICH
Process. At Step 4 of the Process the final draft is recommended for adoption to the
regulatory bodies of the European Union, Japan and USA.
Q6A
Document History
First
Codification
History
Date
New
Codification
November
2005
Q6A
Approval by the Steering Committee under Step 2
and release for public consultation.
18
July
1997
Q6A
Current Step 4 version
Q6A
Approval by the Steering Committee under Step 4 and
recommendation for adoption to the three ICH
regulatory bodies.
6
October
1999
Q6A
i
SPECIFICATIONS: TEST PROCEDURES AND ACCEPTANCE CRITERIA
FOR NEW DRUG SUBSTANCES AND NEW DRUG PRODUCTS:
CHEMICAL SUBSTANCES
ICH Harmonised Tripartite Guideline
Having reached Step 4 of the ICH Process at the ICH Steering Committee meeting
on 6 October 1999, this guideline is recommended for
adoption to the three regulatory parties to ICH
TABLE OF CONTENTS
1. INTRODUCTION ................................................................................................... 1
1.1 Objective of the Guideline ........................................................................... 1
1.2 Background .................................................................................................. 1
1.3 Scope of the Guideline ................................................................................. 1
2. GENERAL CONCEPTS ........................................................................................ 2
2.1 Periodic or Skip Testing ............................................................................... 2
2.2 Release vs. Shelf-life Acceptance Criteria ................................................... 2
2.3 In-process Tests ........................................................................................... 3
2.4 Design and Development Considerations ................................................... 3
2.5 Limited Data Available at Filing................................................................. 3
2.6 Parametric Release ...................................................................................... 4
2.7 Alternative Procedures ................................................................................ 4
2.8 Pharmacopoeial Tests and Acceptance Criteria ......................................... 4
2.9 Evolving Technologies .................................................................................. 5
2.10 Impact of Drug Substance on Drug Product Specifications ....................... 5
2.11 Reference Standard ...................................................................................... 5
3. GUIDELINES ......................................................................................................... 5
3.1 Specifications: Definition and Justification ................................................ 5
3.1.1 Definition of Specifications .......................................................... 5
3.1.2 Justification of Specifications ...................................................... 6

Specifications: New Chemical Drug Substances and Products
ii
3.2 Universal Tests / Criteria ............................................................................. 6
3.2.1 New Drug Substances ................................................................... 6
3.2.2 New Drug Products ....................................................................... 7
3.3 Specific Tests / Criteria ................................................................................ 8
3.3.1 New Drug Substances ................................................................... 8
3.3.2 New Drug Products ..................................................................... 10
4. GLOSSARY ........................................................................................................... 18
5. REFERENCES ...................................................................................................... 20
6. ATTACHMENTS .................................................................................................. 21
1
SPECIFICATIONS: TEST PROCEDURES AND ACCEPTANCE CRITERIA
FOR NEW DRUG SUBSTANCES AND NEW DRUG PRODUCTS:
CHEMICAL SUBSTANCES
1. INTRODUCTION
1.1 Objective of the Guideline
This guideline is intended to assist to the extent possible, in the establishment of a
single set of global specifications for new drug substances and new drug products. It
provides guidance on the setting and justification of acceptance criteria and the
selection of test procedures for new drug substances of synthetic chemical origin, and
new drug products produced from them, which have not been registered previously in
the United States, the European Union, or Japan.
1.2 Background
A specification is defined as a list of tests, references to analytical procedures, and
appropriate acceptance criteria, which are numerical limits, ranges, or other criteria
for the tests described. It establishes the set of criteria to which a drug substance or
drug product should conform to be considered acceptable for its intended use.
"Conformance to specifications" means that the drug substance and / or drug product,
when tested according to the listed analytical procedures, will meet the listed
acceptance criteria. Specifications are critical quality standards that are proposed
and justified by the manufacturer and approved by regulatory authorities as
conditions of approval.
Specifications are one part of a total control strategy for the drug substance and drug
product designed to ensure product quality and consistency. Other parts of this
strategy include thorough product characterization during development, upon which
specifications are based, and adherence to Good Manufacturing Practices; e.g.,
suitable facilities, a validated manufacturing process, validated test procedure, raw
material testing, in-process testing, stability testing, etc.
Specifications are chosen to confirm the quality of the drug substance and drug
product rather than to establish full characterization, and should focus on those
characteristics found to be useful in ensuring the safety and efficacy of the drug
substance and drug product.
1.3 Scope of the Guideline
The quality of drug substances and drug products is determined by their design,
development, in-process controls, GMP controls, and process validation, and by
specifications applied to them throughout development and manufacture. This
guideline addresses specifications, i.e., those tests, procedures, and acceptance
criteria which play a major role in assuring the quality of the new drug substance
and new drug product at release and during shelf life. Specifications are an important
component of quality assurance, but are not its only component. All of the
considerations listed above are necessary to ensure consistent production of drug
substances and drug products of high quality.
This guideline addresses only the marketing approval of new drug products
(including combination products) and, where applicable, new drug substances; it does
not address drug substances or drug products during the clinical research stages of
drug development. This guideline may be applicable to synthetic and semi-synthetic
antibiotics and synthetic peptides of low molecular weight; however, it is not

Specifications: New Chemical Drug Substances and Products
2
sufficient to adequately describe specifications of higher molecular weight peptides
and polypeptides, and biotechnological/biological products. The ICH Guideline
Specifications: Test Procedures and Acceptance Criteria for
Biotechnological/Biological Products addresses guideline specifications, tests and
procedures for biotechnological/biological products. Radiopharmaceuticals, products
of fermentation, oligonucleotides, herbal products and crude products of animal or
plant origin are similarly not covered.
Guidance is provided with regard to acceptance criteria which should be established
for all new drug substances and new drug products, i.e. universal acceptance criteria,
and those that are considered specific to individual drug substances and / or dosage
forms. This guideline should not be considered all encompassing. New analytical
technologies, and modifications to existing technology, are continually being
developed. Such technologies should be used when justified.
Dosage forms addressed in this guideline include solid oral dosage forms, liquid oral
dosage forms, and parenterals (small and large volume). This is not meant to be an
all-inclusive list, or to limit the number of dosage forms to which this guideline
applies. The dosage forms presented serve as models, which may be applicable to
other dosage forms which have not been discussed. The extended application of the
concepts in this guideline to other dosage forms, e.g., to inhalation dosage forms
(powders, solutions, etc.), to topical formulations (creams, ointments, gels), and to
transdermal systems, is encouraged.
2. GENERAL CONCEPTS
The following concepts are important in the development and setting of harmonized
specifications. They are not universally applicable, but each should be considered in
particular circumstances. This guideline presents a brief definition of each concept
and an indication of the circumstances under which it may be applicable. Generally,
proposals to implement these concepts should be justified by the applicant and
approved by the appropriate regulatory authority before being put into effect.
2.1 Periodic or Skip Testing
Periodic or skip testing is the performance of specified tests at release on pre-selected
batches and / or at predetermined intervals, rather than on a batch-to-batch basis
with the understanding that those batches not being tested still must meet all
acceptance criteria established for that product. This represents a less than full
schedule of testing and should therefore be justified and presented to and approved
by the regulatory authority prior to implementation. This concept may be applicable
to, for example, residual solvents and microbiological testing, for solid oral dosage
forms. It is recognized that only limited data may be available at the time of
submission of an application (see section 2.5). This concept should therefore generally
be implemented post-approval. When tested, any failure to meet acceptance criteria
established for the periodic test should be handled by proper notification of the
appropriate regulatory authority(ies). If these data demonstrate a need to restore
routine testing, then batch by batch release testing should be reinstated.
2.2 Release vs. Shelf-life Acceptance Criteria
The concept of different acceptance criteria for release vs. shelf-life specifications
applies to drug products only; it pertains to the establishment of more restrictive
criteria for the release of a drug product than are applied to the shelf-life. Examples
where this may be applicable include assay and impurity (degradation product)
levels. In Japan and the United States, this concept may only be applicable to in-
house criteria, and not to the regulatory release criteria. Thus, in these regions, the

Specifications: New Chemical Drug Substances and Products
3
regulatory acceptance criteria are the same from release throughout shelf-life;
however, an applicant may choose to have tighter in-house limits at the time of
release to provide increased assurance to the applicant that the product will remain
within the regulatory acceptance criterion throughout its shelf-life. In the European
Union there is a regulatory requirement for distinct specifications for release and for
shelf-life where different.
2.3 In-process Tests
In-process tests, as presented in this guideline, are tests which may be performed
during the manufacture of either the drug substance or drug product, rather than as
part of the formal battery of tests which are conducted prior to release.
In-process tests which are only used for the purpose of adjusting process parameters
within an operating range, e.g., hardness and friability of tablet cores which will be
coated and individual tablet weights, are not included in the specification.
Certain tests conducted during the manufacturing process, where the acceptance
criterion is identical to or tighter than the release requirement, (e.g., pH of a
solution) may be sufficient to satisfy specification requirements when the test is
included in the specification. However, this approach should be validated to show
that test results or product performance characteristics do not change from the in-
process stage to finished product.
2.4 Design and Development Considerations
The experience and data accumulated during the development of a new drug
substance or product should form the basis for the setting of specifications. It may be
possible to propose excluding or replacing certain tests on this basis. Some examples
are:
microbiological testing for drug substances and solid dosage forms which have
been shown during development not to support microbial viability or growth
(see Decision Trees #6 and #8);
extractables from product containers where it has been reproducibly shown
that either no extractables are found in the drug product or the levels meet
accepted standards for safety;
particle size testing may fall into this category, may be performed as an in-
process test, or may be performed as a release test, depending on its relevance
to product performance;
dissolution testing for immediate release solid oral drug products made from
highly water soluble drug substances may be replaced by disintegration
testing, if these products have been demonstrated during development to
have consistently rapid drug release characteristics (see Decision Trees #7(1)
through #7(2)).
2.5 Limited Data Available at Filing
It is recognized that only a limited amount of data may be available at the time of
filing, which can influence the process of setting acceptance criteria. As a result it
may be necessary to propose revised acceptance criteria as additional experience is
gained with the manufacture of a particular drug substance or drug product
(example: acceptance limits for a specific impurity). The basis for the acceptance
criteria at the time of filing should necessarily focus on safety and efficacy.
When only limited data are available, the initially approved tests and acceptance
criteria should be reviewed as more information is collected, with a view towards

Specifications: New Chemical Drug Substances and Products
4
possible modification. This could involve loosening, as well as tightening, acceptance
criteria as appropriate.
2.6 Parametric Release
Parametric release can be used as an operational alternative to routine release
testing for the drug product in certain cases when approved by the regulatory
authority. Sterility testing for terminally sterilized drug products is one example. In
this case, the release of each batch is based on satisfactory results from monitoring
specific parameters, e.g., temperature, pressure, and time during the terminal
sterilization phase(s) of drug product manufacturing. These parameters can generally
be more accurately controlled and measured, so that they are more reliable in
predicting sterility assurance than is end-product sterility testing. Appropriate
laboratory tests (e.g., chemical or physical indicator) may be included in the
parametric release program. It is important to note that the sterilization process
should be adequately validated before parametric release is proposed and
maintenance of a validated state should be demonstrated by revalidation at
established intervals. When parametric release is performed, the attribute which is
indirectly controlled (e.g., sterility), together with a reference to the associated test
procedure, still should be included in the specifications.
2.7 Alternative Procedures
Alternative procedures are those which may be used to measure an attribute when
such procedures control the quality of the drug substance or drug product to an
extent that is comparable or superior to the official procedure. Example: for tablets
that have been shown not to degrade during manufacture, it may be permissible to
use a spectrophotometric procedure for release as opposed to the official procedure,
which is chromatographic. However, the chromatographic procedure should still be
used to demonstrate compliance with the acceptance criteria during the shelf-life of
the product.
2.8 Pharmacopoeial Tests and Acceptance Criteria
References to certain procedures are found in pharmacopoeias in each region.
Wherever they are appropriate, pharmacopoeial procedures should be utilized.
Whereas differences in pharmacopoeial procedures and/or acceptance criteria have
existed among the regions, a harmonized specification is possible only if the
procedures and acceptance criteria defined are acceptable to regulatory authorities in
all regions.
The full utility of this Guideline is dependent on the successful completion of
harmonization of pharmacopoeial procedures for several attributes commonly
considered in the specification for new drug substances or new drug products. The
Pharmacopoeial Discussion Group (PDG) of the European Pharmacopoeia, the
Japanese Pharmacopoeia, and the United States Pharmacopeia has expressed a
commitment to achieving harmonization of the procedures in a timely fashion.
Where harmonization has been achieved, an appropriate reference to the harmonized
procedure and acceptance criteria is considered acceptable for a specification in all
three regions. For example, after harmonization sterility data generated using the
JP procedure, as well as the JP procedure itself and its acceptance criteria, are
considered acceptable for registration in all three regions. To signify the harmonized
status of these procedures, the pharmacopoeias have agreed to include a statement in
their respective texts which indicates that the procedures and acceptance criteria
from all three pharmacopoeias are considered equivalent and are, therefore,
interchangeable.

Specifications: New Chemical Drug Substances and Products
5
Since the overall value of this Guideline is linked to the extent of harmonization of
the analytical procedures and acceptance criteria of the pharmacopoeias, it is agreed
by the members of the Q6A expert working group that none of the three
pharmacopoeias should change a harmonized monograph unilaterally. According to
the PDG procedure for the revision of harmonized monographs and chapters, “no
pharmacopoeia shall revise unilaterally any monograph or chapter after sign-off or
after publication.”
2.9 Evolving Technologies
New analytical technologies, and modifications to existing technology, are continually
being developed. Such technologies should be used when they are considered to offer
additional assurance of quality, or are otherwise justified.
2.10 Impact of Drug Substance on Drug Product Specifications
In general, it should not be necessary to test the drug product for quality attributes
uniquely associated with the drug substance. Example: it is normally not considered
necessary to test the drug product for synthesis impurities which are controlled in the
drug substance and are not degradation products. Refer to the ICH Guideline
Impurities in New Drug Products for detailed information.
2.11 Reference Standard
A reference standard, or reference material, is a substance prepared for use as the
standard in an assay, identification, or purity test. It should have a quality
appropriate to its use. It is often characterized and evaluated for its intended purpose
by additional procedures other than those used in routine testing. For new drug
substance reference standards intended for use in assays, the impurities should be
adequately identified and / or controlled, and purity should be measured by a
quantitative procedure.
3. GUIDELINES
3.1 Specifications: Definition and Justification
3.1.1 Definition of Specifications
A specification is defined as a list of tests, references to analytical procedures, and
appropriate acceptance criteria which are numerical limits, ranges, or other criteria
for the tests described. It establishes the set of criteria to which a new drug
substance or new drug product should conform to be considered acceptable for its
intended use. "Conformance to specifications" means that the drug substance and / or
drug product, when tested according to the listed analytical procedures, will meet the
listed acceptance criteria. Specifications are critical quality standards that are
proposed and justified by the manufacturer and approved by regulatory authorities
as conditions of approval.
It is possible that, in addition to release tests, a specification may list in-process tests
as defined in 2.3, periodic (skip) tests, and other tests which are not always
conducted on a batch-by-batch basis. In such cases the applicant should specify which
tests are routinely conducted batch-by-batch, and which tests are not, with an
indication and justification of the actual testing frequency. In this situation, the drug
substance and / or drug product should meet the acceptance criteria if tested.
It should be noted that changes in the specification after approval of the application
may need prior approval by the regulatory authority.

Specifications: New Chemical Drug Substances and Products
6
3.1.2 Justification of Specifications
When a specification is first proposed, justification should be presented for each
procedure and each acceptance criterion included. The justification should refer to
relevant development data, pharmacopoeial standards, test data for drug substances
and drug products used in toxicology and clinical studies, and results from
accelerated and long term stability studies, as appropriate. Additionally, a reasonable
range of expected analytical and manufacturing variability should be considered. It is
important to consider all of this information.
Approaches other than those set forth in this guideline may be applicable and
acceptable. The applicant should justify alternative approaches. Such justification
should be based on data derived from the new drug substance synthesis and/or the
new drug product manufacturing process. This justification may consider theoretical
tolerances for a given procedure or acceptance criterion, but the actual results
obtained should form the primary basis for whatever approach is taken.
Test results from stability and scale-up / validation batches, with emphasis on the
primary stability batches, should be considered in setting and justifying
specifications. If multiple manufacturing sites are planned, it may be valuable to
consider data from these sites in establishing the initial tests and acceptance criteria.
This is particularly true when there is limited initial experience with the
manufacture of the drug substance or drug product at any particular site. If data
from a single representative manufacturing site are used in setting tests and
acceptance criteria, product manufactured at all sites should still comply with these
criteria.
Presentation of test results in graphic format may be helpful in justifying individual
acceptance criteria, particularly for assay values and impurity levels. Data from
development work should be included in such a presentation, along with stability
data available for new drug substance or new drug product batches manufactured by
the proposed commercial processes. Justification for proposing exclusion of a test
from the specification should be based on development data and on process validation
data (where appropriate).
3.2 Universal Tests / Criteria
Implementation of the recommendations in the following section should take into
account the ICH Guidelines “Text on Validation of Analytical Procedures” and
“Validation of Analytical Procedures: Methodology”.
3.2.1 New Drug Substances
The following tests and acceptance criteria are considered generally applicable to all
new drug substances.
a) Description: a qualitative statement about the state (e.g. solid, liquid) and color of
the new drug substance. If any of these characteristics change during storage, this
change should be investigated and appropriate action taken.
b) Identification: identification testing should optimally be able to discriminate
between compounds of closely related structure which are likely to be present.
Identification tests should be specific for the new drug substance, e.g., infrared
spectroscopy. Identification solely by a single chromatographic retention time, for
example, is not regarded as being specific. However, the use of two chromatographic
procedures, where the separation is based on different principles or a combination of
tests into a single procedure, such as HPLC/UV diode array, HPLC/MS, or GC/MS is
generally acceptable. If the new drug substance is a salt, identification testing should

Specifications: New Chemical Drug Substances and Products
7
be specific for the individual ions. An identification test that is specific for the salt
itself should suffice.
New drug substances which are optically active may also need specific identification
testing or performance of a chiral assay. Please refer to 3.3.1.d) in this Guideline for
further discussion of this topic.
c) Assay: A specific, stability-indicating procedure should be included to determine
the content of the new drug substance. In many cases it is possible to employ the
same procedure (e.g., HPLC) for both assay of the new drug substance and
quantitation of impurities.
In cases where use of a non-specific assay is justified, other supporting analytical
procedures should be used to achieve overall specificity. For example, where titration
is adopted to assay the drug substance, the combination of the assay and a suitable
test for impurities should be used.
d) Impurities: Organic and inorganic impurities and residual solvents are included in
this category. Refer to the ICH Guidelines Impurities in New Drug Substances and
Residual Solvents in Pharmaceuticals for detailed information.
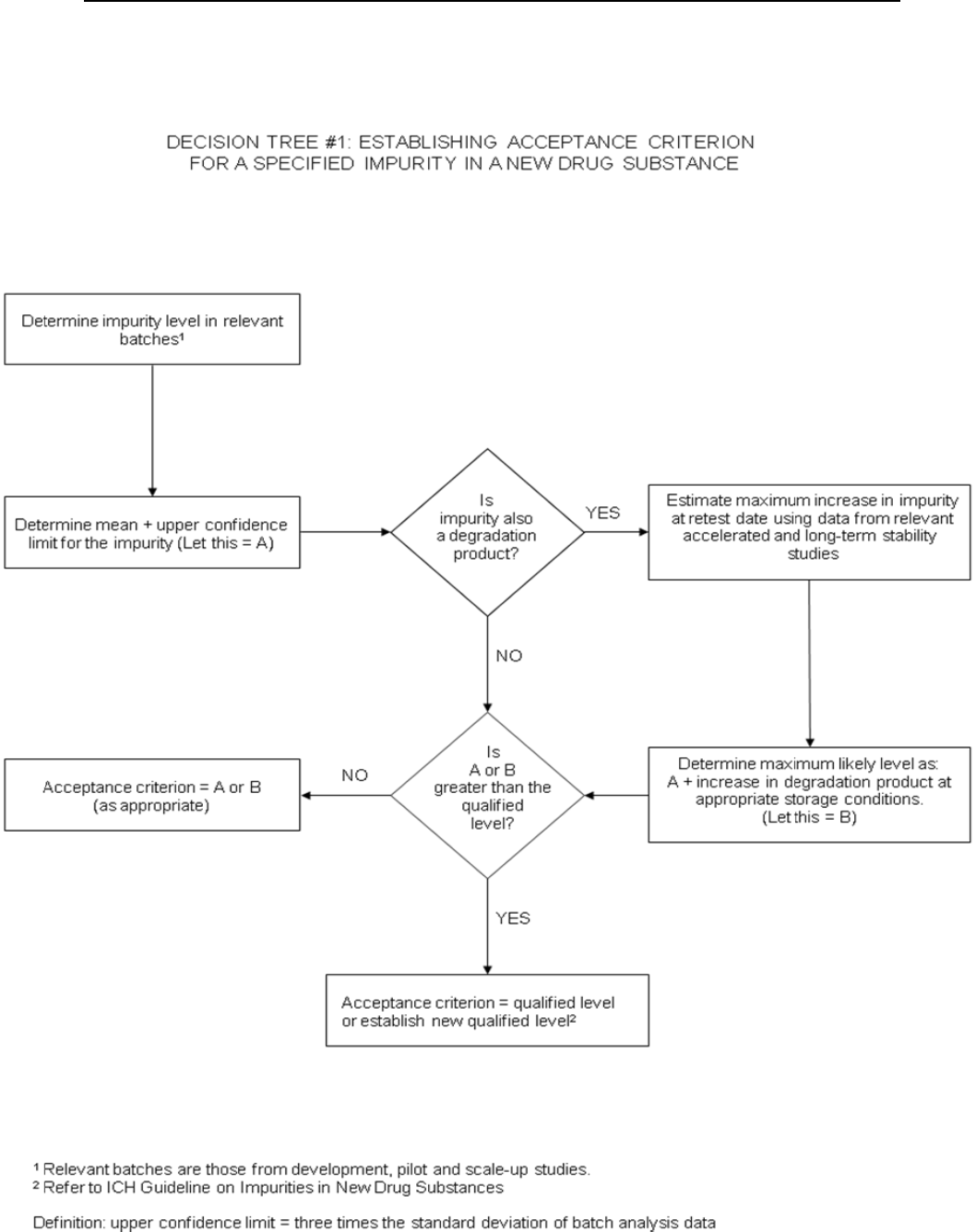
Decision tree #1 addresses the extrapolation of meaningful limits on impurities from
the body of data generated during development. At the time of filing it is unlikely
that sufficient data will be available to assess process consistency. Therefore it is
considered inappropriate to establish acceptance criteria which tightly encompass the
batch data at the time of filing. (see section 2.5)
3.2.2 New Drug Products
The following tests and acceptance criteria are considered generally applicable to all
new drug products:
a) Description: A qualitative description of the dosage form should be provided (e.g.,
size, shape, and color). If any of these characteristics change during manufacture or
storage, this change should be investigated and appropriate action taken. The
acceptance criteria should include the final acceptable appearance. If color changes
during storage, a quantitative procedure may be appropriate.
b) Identification: Identification testing should establish the identity of the new drug
substance(s) in the new drug product and should be able to discriminate between
compounds of closely related structure which are likely to be present. Identity tests
should be specific for the new drug substance, e.g., infrared spectroscopy.
Identification solely by a single chromatographic retention time, for example, is not
regarded as being specific. However, the use of two chromatographic procedures,
where the separation is based on different principles, or combination of tests into a
single procedure, such as HPLC/UV diode array, HPLC/MS, or GC/MS, is generally
acceptable.
c) Assay: A specific, stability-indicating assay to determine strength (content) should
be included for all new drug products. In many cases it is possible to employ the same
procedure (e.g., HPLC) for both assay of the new drug substance and quantitation of
impurities. Results of content uniformity testing for new drug products can be used
for quantitation of drug product strength, if the methods used for content uniformity
are also appropriate as assays.
In cases where use of a non-specific assay is justified, other supporting analytical
procedures should be used to achieve overall specificity. For example, where titration
is adopted to assay the drug substance for release, the combination of the assay and a

Specifications: New Chemical Drug Substances and Products
8
suitable test for impurities can be used. A specific procedure should be used when
there is evidence of excipient interference with the non-specific assay.
d) Impurities: Organic and inorganic impurities (degradation products) and residual
solvents are included in this category. Refer to the ICH Guidelines Impurities in
New Drug Products and Residual Solvents for detailed information.
Organic impurities arising from degradation of the new drug substance and
impurities that arise during the manufacturing process for the drug product should
be monitored in the new drug product. Acceptance limits should be stated for
individual specified degradation products, which may include both identified and
unidentified degradation products as appropriate, and total degradation products.
Process impurities from the new drug substance synthesis are normally controlled
during drug substance testing, and therefore are not included in the total impurities
limit. However, when a synthesis impurity is also a degradation product, its level
should be monitored and included in the total degradation product limit. When it has
been conclusively demonstrated via appropriate analytical methodology, that the
drug substance does not degrade in the specific formulation, and under the specific
storage conditions proposed in the new drug application, degradation product testing
may be reduced or eliminated upon approval by the regulatory authorities.
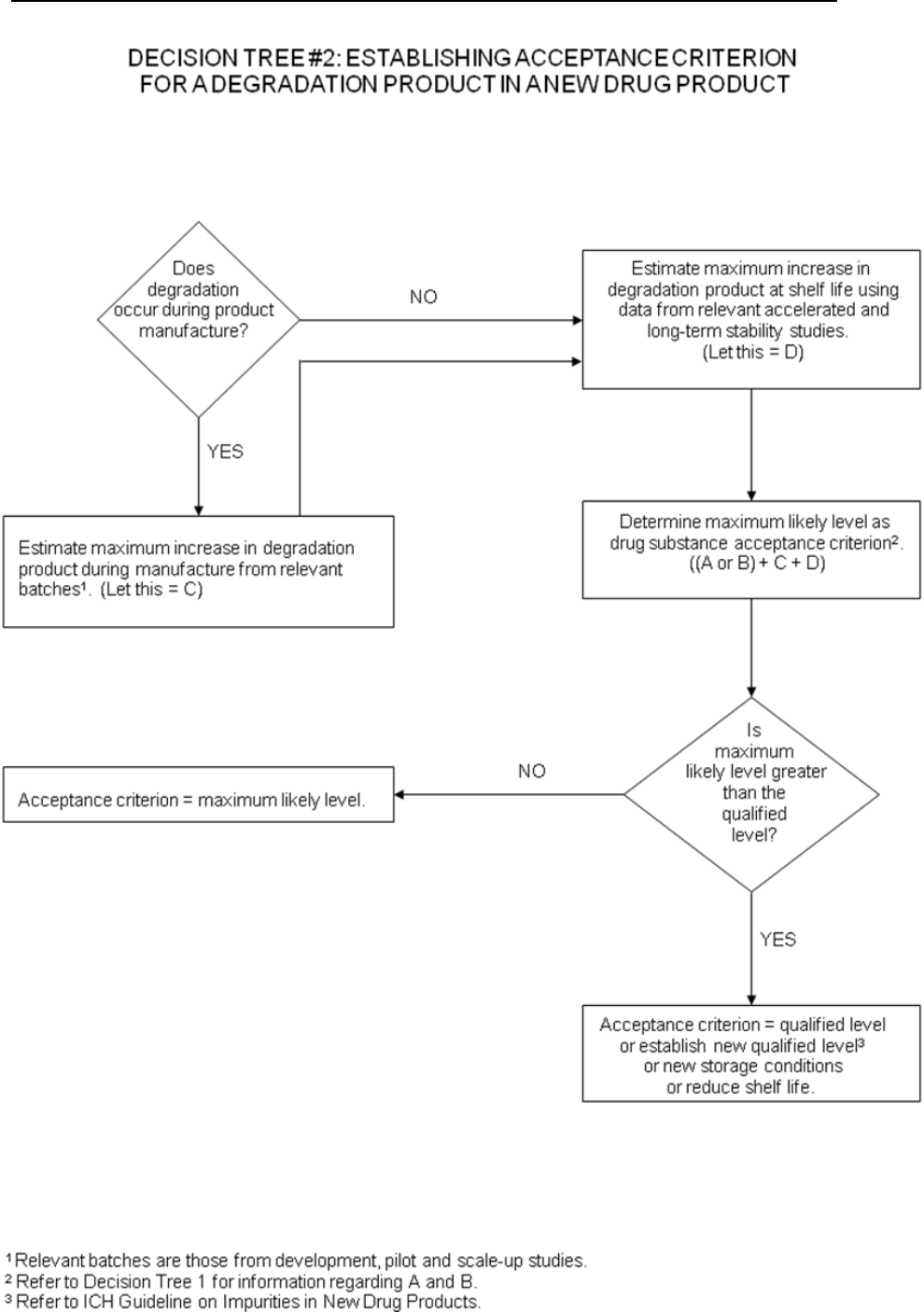
Decision tree #2 addresses the extrapolation of meaningful limits on degradation
products from the body of data generated during development. At the time of filing it
is unlikely that sufficient data will be available to assess process consistency.
Therefore it is considered inappropriate to establish acceptance criteria which tightly
encompass the batch data at the time of filing. (see section 2.5)
3.3 Specific Tests / Criteria
In addition to the universal tests listed above, the following tests may be considered
on a case by case basis for drug substances and/or drug products. Individual
tests/criteria should be included in the specification when the tests have an impact on
the quality of the drug substance and drug product for batch control. Tests other than
those listed below may be needed in particular situations or as new information
becomes available.
3.3.1 New Drug Substances
a) Physicochemical properties: These are properties such as pH of an aqueous
solution, melting point / range, and refractive index. The procedures used for the
measurement of these properties are usually unique and do not need much
elaboration, e.g., capillary melting point, Abbé refractometry. The tests performed in
this category should be determined by the physical nature of the new drug substance
and by its intended use.
b) Particle size: For some new drug substances intended for use in solid or suspension
drug products, particle size can have a significant effect on dissolution rates,
bioavailability, and / or stability. In such instances, testing for particle size
distribution should be carried out using an appropriate procedure, and acceptance
criteria should be provided.
Decision tree #3 provides additional guidance on when particle size testing should be
considered.
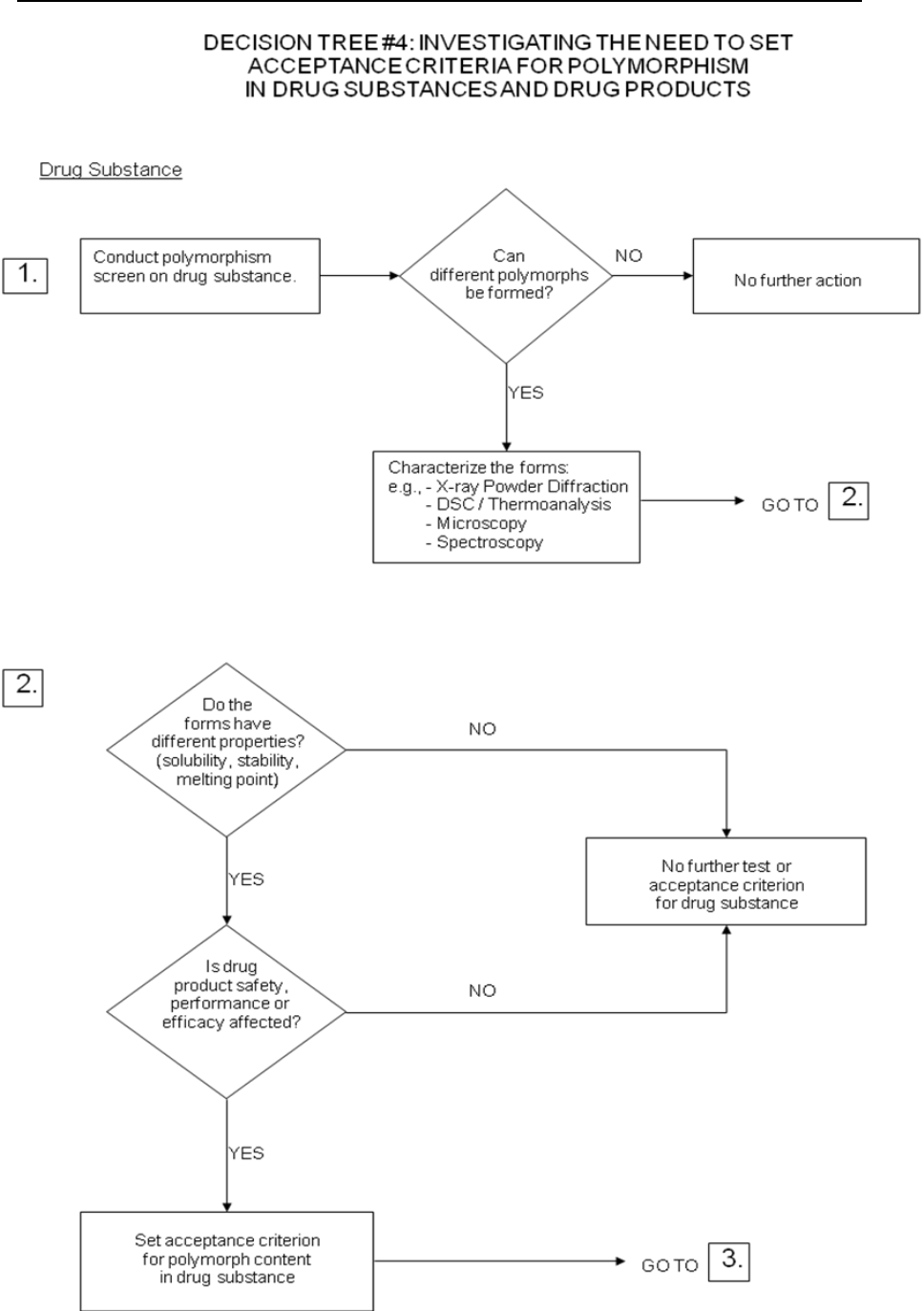
c) Polymorphic forms: Some new drug substances exist in different crystalline forms
which differ in their physical properties. Polymorphism may also include solvation or
hydration products (also known as pseudopolymorphs) and amorphous forms.
Differences in these forms could, in some cases, affect the quality or performance of

Specifications: New Chemical Drug Substances and Products
9
the new drug products. In cases where differences exist which have been shown to
affect drug product performance, bioavailability or stability, then the appropriate
solid state should be specified.
Physicochemical measurements and techniques are commonly used to determine
whether multiple forms exist. Examples of these procedures are: melting point
(including hot-stage microscopy), solid state IR, X-ray powder diffraction, thermal
analysis procedures (like DSC, TGA and DTA), Raman spectroscopy, optical
microscopy, and solid state NMR.
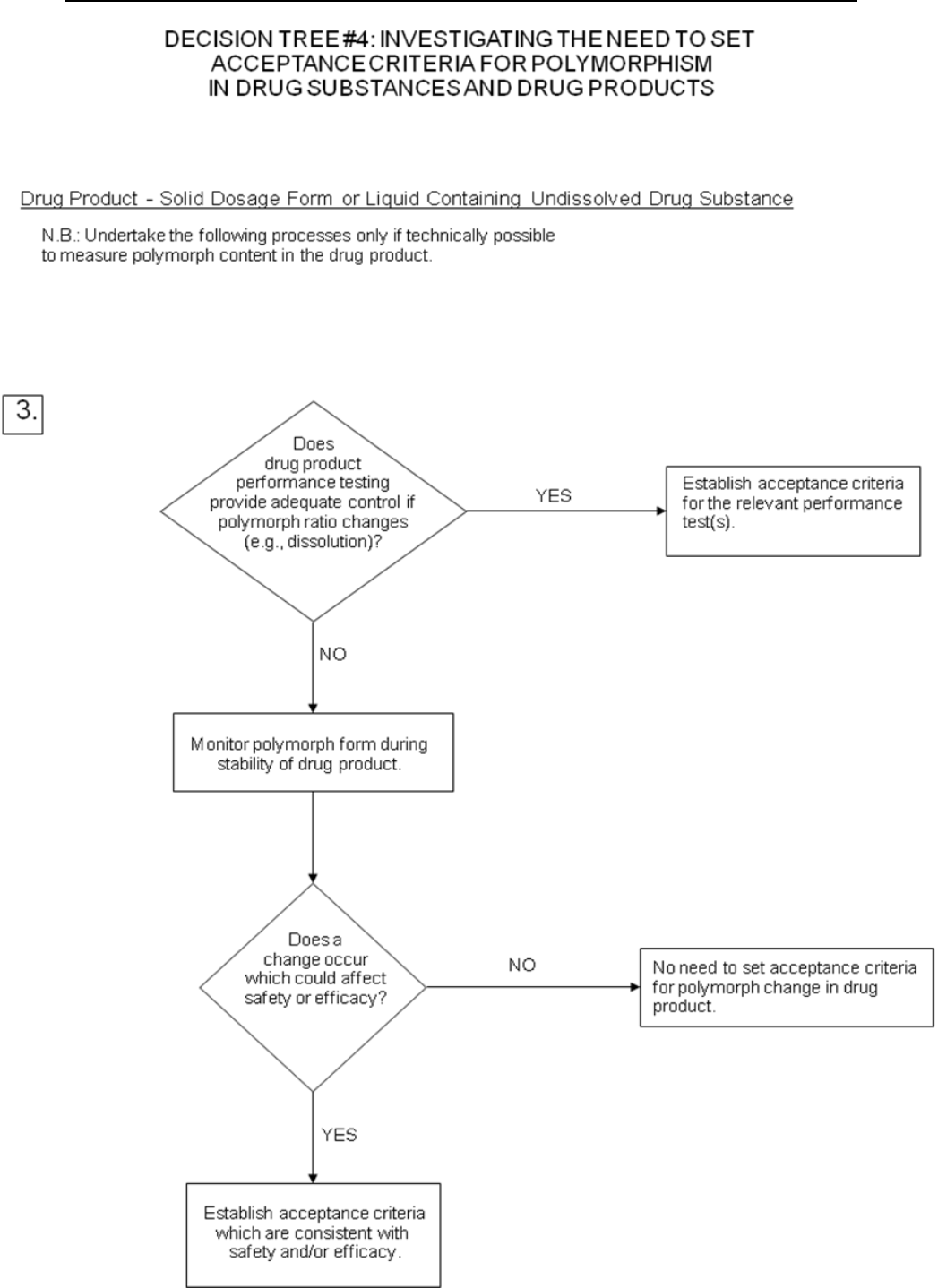
Decision trees #4(1) through 4(3) provide additional guidance on when, and how,
polymorphic forms should be monitored and controlled.
Note: These decision trees should be followed sequentially. Trees 1 and 2 consider
whether polymorphism is exhibited by the drug substance, and whether the different
polymorphic forms can affect performance of the drug product. Tree 3 should only be
applied when polymorphism has been demonstrated for the drug substance, and
shown to affect these properties. Tree 3 considers the potential for change in
polymorphic forms in the drug product, and whether such a change has any effect on
product performance.
It is generally technically very difficult to measure polymorphic changes in drug
products. A surrogate test (e.g., dissolution) (see Decision tree 4(3)) can generally be
used to monitor product performance, and polymorph content should only be used as
a test and acceptance criterion of last resort.
d) Tests for chiral new drug substances: Where a new drug substance is
predominantly one enantiomer, the opposite enantiomer is excluded from the
qualification and identification thresholds given in the ICH Guidelines on Impurities
in New Drug Substances and Impurities in New Drug Products because of practical
difficulties in quantifying it at those levels. However, that impurity in the chiral new
drug substance and the resulting new drug product(s) should otherwise be treated
according to the principles established in those Guidelines.
Decision tree #5 summarizes when and if chiral identity tests, impurity tests, and
assays may be needed for both new drug substances and new drug products,
according to the following concepts:
Drug Substance: Impurities. For chiral drug substances which are developed as a
single enantiomer, control of the other enantiomer should be considered in the
same manner as for other impurities. However, technical limitations may
preclude the same limits of quantification or qualification from being applied.
Assurance of control also could be given by appropriate testing of a starting
material or intermediate, with suitable justification.
Assay. An enantioselective determination of the drug substance should be part of
the specification. It is considered acceptable for this to be achieved either through
use of a chiral assay procedure or by the combination of an achiral assay together
with appropriate methods of controlling the enantiomeric impurity.
Identity. For a drug substance developed as a single enantiomer, the identity
test(s) should be capable of distinguishing both enantiomers and the racemic
mixture. For a racemic drug substance, there are generally two situations where
a stereospecific identity test is appropriate for release/acceptance testing: 1)
where there is a significant possibility that the enantiomer might be substituted
for the racemate, or 2) when there is evidence that preferential crystallization
may lead to unintentional production of a non-racemic mixture.

Specifications: New Chemical Drug Substances and Products
10
Drug Product: Degradation products. Control of the other enantiomer in a drug
product is considered necessary unless racemization has been shown to be
insignificant during manufacture of the dosage form, and on storage.
Assay: An achiral assay may be sufficient where racemization has been shown to
be insignificant during manufacture of the dosage form, and on storage.
Otherwise a chiral assay should be used, or alternatively, the combination of an
achiral assay plus a validated procedure to control the presence of the opposite
enantiomer may be used.
Identity: A stereospecific identity test is not generally needed in the drug product
release specification. When racemization is insignificant during manufacture of
the dosage form, and on storage, stereospecific identity testing is more
appropriately addressed as part of the drug substance specification. When
racemization in the dosage form is a concern, chiral assay or enantiomeric
impurity testing of the drug product will serve to verify identity.
e) Water content: This test is important in cases where the new drug substance is
known to be hygroscopic or degraded by moisture or when the drug substance is
known to be a stoichiometric hydrate. The acceptance criteria may be justified with
data on the effects of hydration or moisture absorption. In some cases, a Loss on
Drying procedure may be considered adequate; however, a detection procedure that is
specific for water (e.g., Karl Fischer titration) is preferred.
f) Inorganic impurities: The need for inclusion of tests and acceptance criteria for
inorganic impurities (e.g., catalysts) should be studied during development and based
on knowledge of the manufacturing process. Procedures and acceptance criteria for
sulfated ash / residue on ignition should follow pharmacopoeial precedents; other
inorganic impurities may be determined by other appropriate procedures, e.g., atomic
absorption spectroscopy.
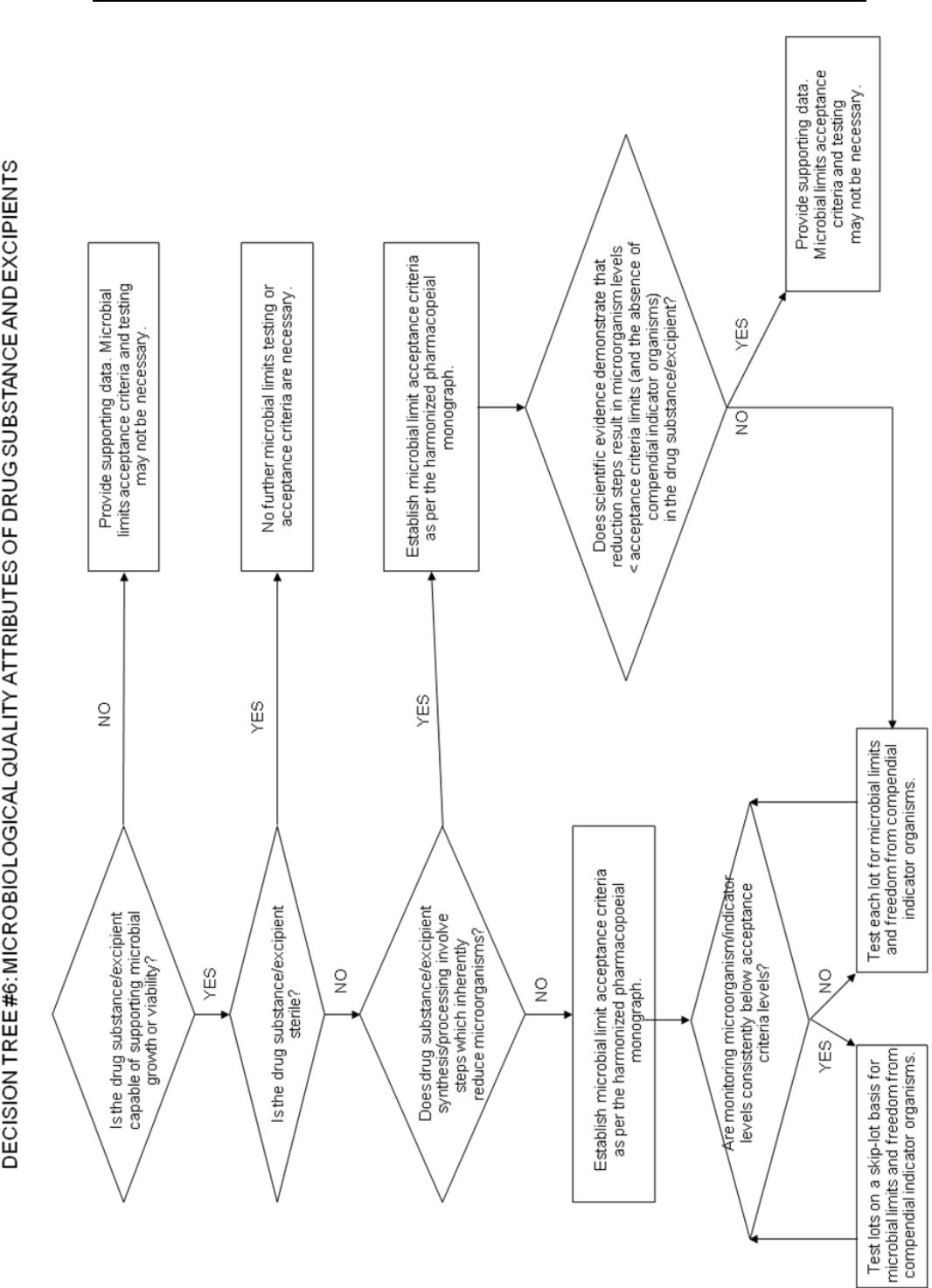
g) Microbial limits: There may be a need to specify the total count of aerobic
microorganisms, the total count of yeasts and molds, and the absence of specific
objectionable bacteria (e.g., Staphylococcus aureus, Escherichia coli, Salmonella,
Pseudomonas aeruginosa). These should be suitably determined using
pharmacopoeial procedures. The type of microbial test(s) and acceptance criteria
should be based on the nature of the drug substance, method of manufacture, and the
intended use of the drug product. For example, sterility testing may be appropriate
for drug substances manufactured as sterile and endotoxin testing may be
appropriate for drug substances used to formulate an injectable drug product.
Decision tree #6 provides additional guidance on when microbial limits should be
included.
3.3.2 New Drug Products
Additional tests and acceptance criteria generally should be included for particular
new drug products. The following selection presents a representative sample of both
the drug products and the types of tests and acceptance criteria which may be
appropriate. The specific dosage forms addressed include solid oral drug products,
liquid oral drug products, and parenterals (small and large volume). Application of
the concepts in this guideline to other dosage forms is encouraged. Note that issues
related to optically active drug substances and to solid state considerations for drug
products are discussed in part 3.3.1. of this guideline.
3.3.2.1 The following tests are applicable to tablets (coated and uncoated) and hard
capsules. One or more of these tests may also be applicable to soft capsules and
granules.

Specifications: New Chemical Drug Substances and Products
11
a) Dissolution: The specification for solid oral dosage forms normally includes a test
to measure release of drug substance from the drug product. Single-point
measurements are normally considered to be suitable for immediate-release dosage
forms. For modified-release dosage forms, appropriate test conditions and sampling
procedures should be established. For example, multiple time point sampling should
be performed for extended-release dosage forms, and two-stage testing (using
different media in succession or in parallel, as appropriate) may be appropriate for
delayed-release dosage forms. In these cases it is important to consider the
populations of individuals who will be taking the drug product (e.g., achlorhydric
elderly) when designing the tests and acceptance criteria. In some cases (see 3.3.2.1
b) Disintegration) dissolution testing may be replaced by disintegration testing (see
Decision Tree #7 (1)).
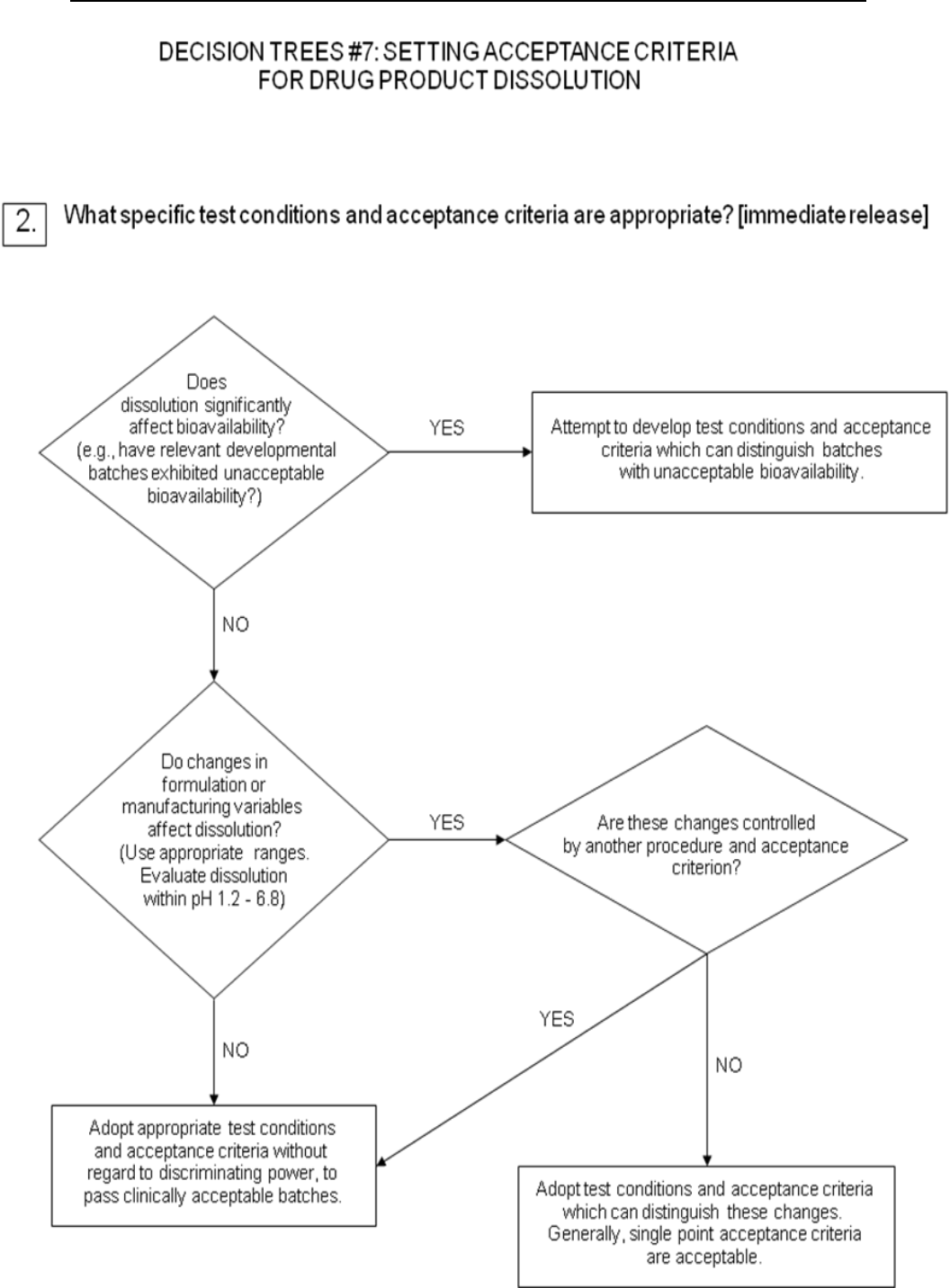
For immediate-release drug products where changes in dissolution rate have been
demonstrated to significantly affect bioavailability, it is desirable to develop test
conditions which can distinguish batches with unacceptable bioavailability. If
changes in formulation or process variables significantly affect dissolution and such
changes are not controlled by another aspect of the specification, it may also be
appropriate to adopt dissolution test conditions which can distinguish these changes
(see Decision Tree #7(2)).
Where dissolution significantly affects bioavailability, the acceptance criteria should
be set to reject batches with unacceptable bioavailability. Otherwise, test conditions
and acceptance criteria should be established which pass clinically acceptable batches
(see Decision Tree #7(2)).
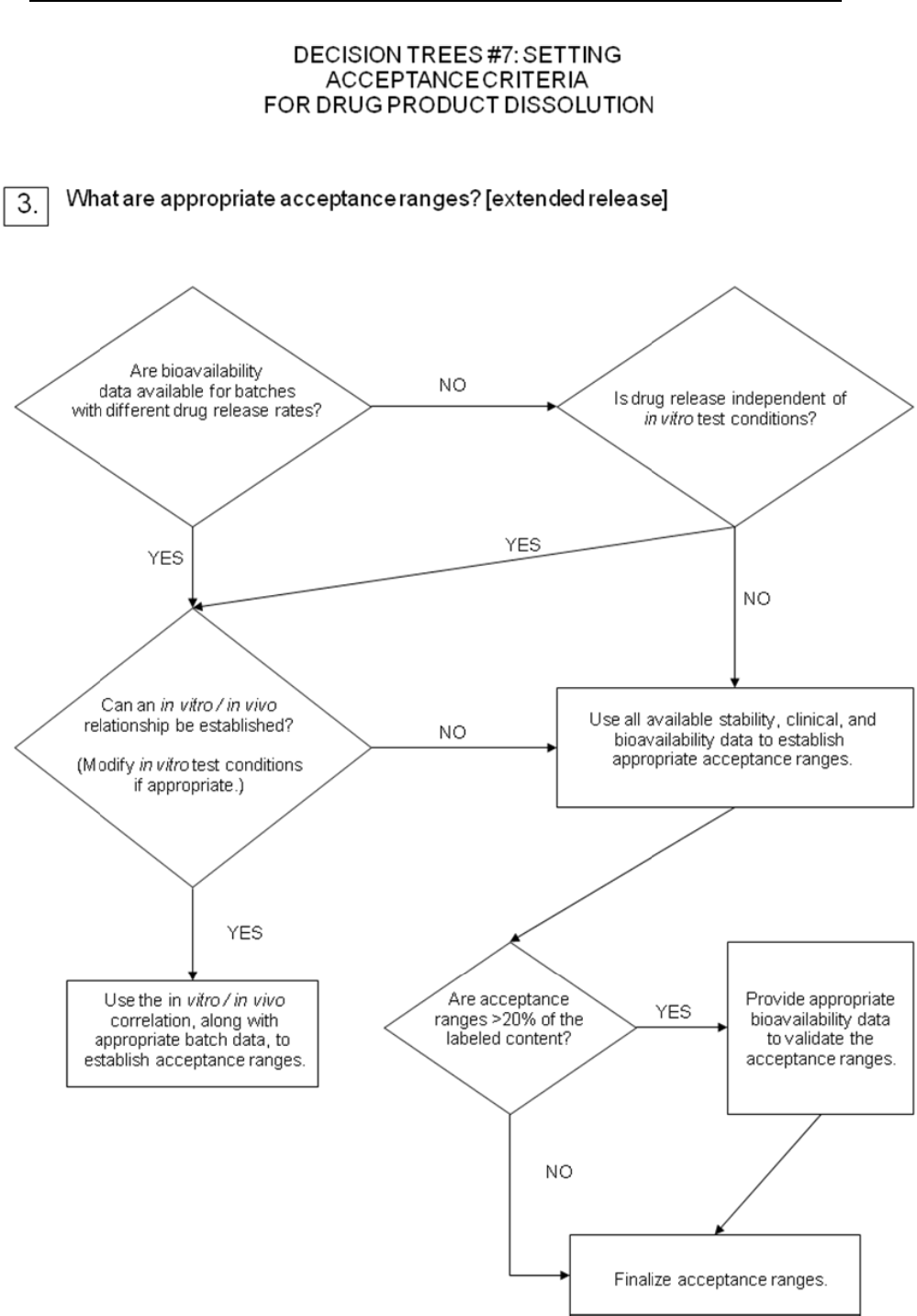
For extended-release drug products, in vitro / in vivo correlation may be used to
establish acceptance criteria when human bioavailability data are available for
formulations exhibiting different release rates. Where such data are not available,
and drug release cannot be shown to be independent of in vitro test conditions, then
acceptance criteria should be established on the basis of available batch data.
Normally, the permitted variability in mean release rate at any given time point
should not exceed a total numerical difference of +/-10% of the labeled content of drug
substance (i.e., a total variability of 20%: a requirement of 50 +/- 10% thus means an
acceptable range from 40% to 60%), unless a wider range is supported by a
bioequivalency study (see Decision Tree #7(3)).
b) Disintegration: For rapidly dissolving (dissolution >80% in 15 minutes at pH 1.2,
4.0 and 6.8) products containing drugs which are highly soluble throughout the
physiological range (dose/solubility volume < 250 mL from pH 1.2 to 6.8),
disintegration may be substituted for dissolution. Disintegration testing is most
appropriate when a relationship to dissolution has been established or when
disintegration is shown to be more discriminating than dissolution. In such cases
dissolution testing may not be necessary. It is expected that development information
will be provided to support the robustness of the formulation and manufacturing
process with respect to the selection of dissolution vs. disintegration testing (see
Decision Tree #7(1)).
c) Hardness/friability: It is normally appropriate to perform hardness and/or
friability testing as an in-process control (see section 2.3). Under these circumstances,
it is normally not necessary to include these attributes in the specification. If the
characteristics of hardness and friability have a critical impact on drug product
quality (e.g., chewable tablets), acceptance criteria should be included in the
specification.

Specifications: New Chemical Drug Substances and Products
12
d) Uniformity of dosage units: This term includes both the mass of the dosage form
and the content of the active substance in the dosage form; a pharmacopoeial
procedure should be used. In general, the specification should include one or the
other but not both. If appropriate, these tests may be performed in-process; the
acceptance criteria should be included in the specification. When weight variation is
applied for new drug products exceeding the threshold value to allow testing
uniformity by weight variation, applicants should verify during drug development
that the homogeneity of the product is adequate.
e) Water content: A test for water content should be included when appropriate. The
acceptance criteria may be justified with data on the effects of hydration or water
absorption on the drug product. In some cases, a Loss on Drying procedure may be
considered adequate; however, a detection procedure which is specific for water (e.g.,
Karl Fischer titration) is preferred.
f) Microbial limits: Microbial limit testing is seen as an attribute of Good
Manufacturing Practice, as well as of quality assurance. In general, it is advisable to
test the drug product unless its components are tested before manufacture and the
manufacturing process is known, through validation studies, not to carry a
significant risk of microbial contamination or proliferation. It should be noted that,
whereas this guideline does not directly address excipients, the principles discussed
here may be applicable to excipients as well as to new drug products. Skip testing
may be an appropriate approach in both cases where permissible. (See Decision Tree
#6 for microbial testing of excipients.)
Acceptance criteria should be set for the total count of aerobic microorganisms, the
total count of yeasts and molds, and the absence of specific objectionable bacteria
(e.g., Staphylococcus aureus, Escherichia coli, Salmonella, Pseudomonas aeruginosa).
These should be determined by suitable procedures, using pharmacopoeial
procedures, and at a sampling frequency or time point in manufacture which is
justified by data and experience. The type of microbial test(s) and acceptance criteria
should be based on the nature of the drug substance, method of manufacture, and the
intended use of the drug product. With acceptable scientific justification, it should be
possible to propose no microbial limit testing for solid oral dosage forms.
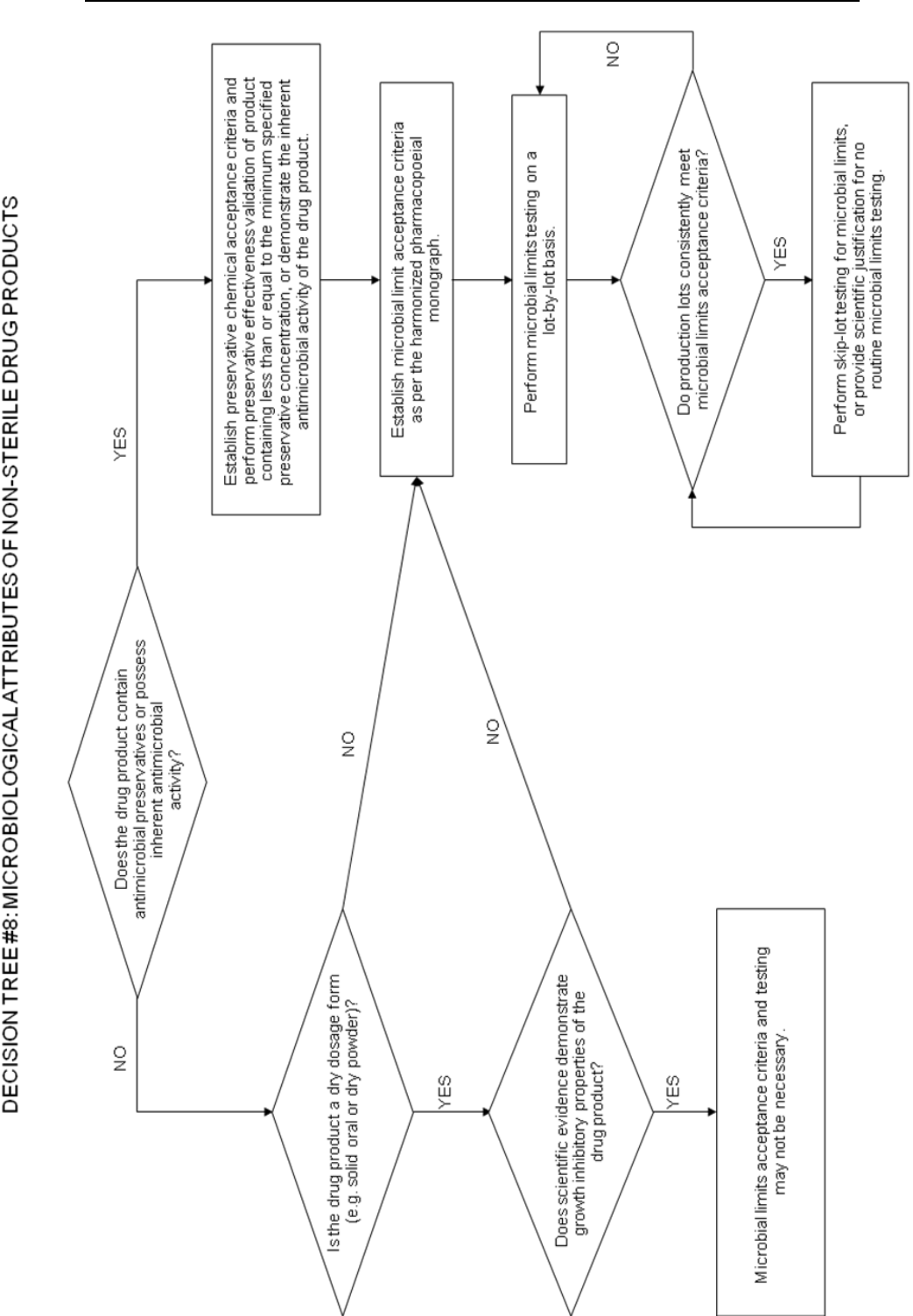
Decision tree #8 provides additional guidance on the use of microbial limits testing.
3.3.2.2 Oral liquids: One or more of the following specific tests will normally be
applicable to oral liquids and to powders intended for reconstitution as oral liquids.
a) Uniformity of dosage units: This term includes both the mass of the dosage form
and the content of the active substance in the dosage form; a pharmacopoeial
procedure should be used. In general, the specification should include one or the
other but not both. When weight variation is applied for new drug products
exceeding the threshold value to allow testing uniformity by weight variation,
applicants should verify during drug development that the homogeneity of the
product is adequate.
If appropriate, tests may be performed in-process; however, the acceptance criteria
should be included in the specification. This concept may be applied to both single-
dose and multiple-dose packages.
The dosage unit is considered to be the typical dose taken by the patient. If the actual
unit dose, as taken by the patient, is controlled, it may either be measured directly or
calculated, based on the total measured weight or volume of drug divided by the total
number of doses expected. If dispensing equipment (such as medicine droppers or
dropper tips for bottles) is an integral part of the packaging, this equipment should

Specifications: New Chemical Drug Substances and Products
13
be used to measure the dose. Otherwise, a standard volume measure should be used.
The dispensing equipment to be used is normally determined during development.
For powders for reconstitution, uniformity of mass testing is generally considered
acceptable.
b) pH: Acceptance criteria for pH should be provided where applicable and the
proposed range justified.
c) Microbial limits: Microbial limit testing is seen as an attribute of Good
Manufacturing Practice, as well as of quality assurance. In general, it is advisable to
test the drug product unless its components are tested before manufacture and the
manufacturing process is known, through validation studies, not to carry a
significant risk of microbial contamination or proliferation. It should be noted that,
whereas this Guideline does not directly address excipients, the principles discussed
here may be applicable to excipients as well as to new drug products. Skip testing
may be an appropriate approach in both cases where permissible. With acceptable
scientific justification, it may be possible to propose no microbial limit testing for
powders intended for reconstitution as oral liquids.
Acceptance criteria should be set for the total count of aerobic microorganisms, total
count of yeasts and molds, and the absence of specific objectionable bacteria (e.g.,
Staphylococcus aureus, Escherichia coli, Salmonella, Pseudomonas aeruginosa).
These should be determined by suitable procedures, using pharmacopoeial
procedures, and at a sampling frequency or time point in manufacture which is
justified by data and experience.
Decision tree #8 provides additional guidance on the use of microbial limits testing.
d) Antimicrobial preservative content: For oral liquids needing an antimicrobial
preservative, acceptance criteria for preservative content should be established.
Acceptance criteria for preservative content should be based upon the levels of
antimicrobial preservative necessary to maintain microbiological quality of the
product at all stages throughout its proposed usage and shelf-life. The lowest
specified concentration of antimicrobial preservative should be demonstrated to be
effective in controlling microorganisms by using a pharmacopoeial antimicrobial
preservative effectiveness test.
Testing for antimicrobial preservative content should normally be performed at
release. Under certain circumstances, in-process testing may suffice in lieu of release
testing. When antimicrobial preservative content testing is performed as an in-
process test, the acceptance criteria should remain part of the specification.
Antimicrobial preservative effectiveness should be demonstrated during
development, during scaleup, and throughout the shelf-life (e.g., in stability testing:
see the ICH Guideline, “Stability Testing of New Drug Substances and Products”),
although chemical testing for preservative content is the attribute normally included
in the specification.
e) Antioxidant preservative content: Release testing for antioxidant content should
normally be performed. Under certain circumstances, where justified by
developmental and stability data, shelf-life testing may be unnecessary, and in-
process testing may suffice in lieu of release testing where permitted. When
antioxidant content testing is performed as an in-process test, the acceptance criteria
should remain part of the specification. If only release testing is performed, this
decision should be reinvestigated whenever either the manufacturing procedure or
the container/closure system changes.

Specifications: New Chemical Drug Substances and Products
14
f) Extractables: Generally, where development and stability data show evidence that
extractables from the container/closure systems are consistently below levels that are
demonstrated to be acceptable and safe, elimination of this test can normally be
accepted. This should be reinvestigated if the container/closure system or
formulation changes.
Where data demonstrate the need, tests and acceptance criteria for extractables from
the container/closure system components (e.g., rubber stopper, cap liner, plastic
bottle, etc.) are considered appropriate for oral solutions packaged in non-glass
systems, or in glass containers with non-glass closures. The container/closure
components should be listed, and data collected for these components as early in the
development process as possible.
g) Alcohol content: Where it is declared quantitatively on the label in accordance with
pertinent regulations, the alcohol content should be specified. It may be assayed or
calculated.
h) Dissolution: In addition to the attributes recommended immediately above, it may
be appropriate (e.g., insoluble drug substance) to include dissolution testing and
acceptance criteria for oral suspensions and dry powder products for resuspension.
Dissolution testing should be performed at release. This test may be performed as an
in-process test when justified by product development data. The testing apparatus,
media, and conditions should be pharmacopoeial, if possible, or otherwise justified.
Dissolution procedures using either pharmacopoeial or non-pharmacopoeial
apparatus and conditions should be validated.
Single-point measurements are normally considered suitable for immediate-release
dosage forms. Multiple-point sampling, at appropriate intervals, should be performed
for modified-release dosage forms. Acceptance criteria should be set based on the
observed range of variation, and should take into account the dissolution profiles of
the batches that showed acceptable performance in vivo. Developmental data should
be considered when determining the need for either a dissolution procedure or a
particle size distribution procedure.
i) Particle size distribution: Quantitative acceptance criteria and a procedure for
determination of particle size distribution may be appropriate for oral suspensions.
Developmental data should be considered when determining the need for either a
dissolution procedure or a particle size distribution procedure for these formulations.
Particle size distribution testing should be performed at release. It may be performed
as an in-process test when justified by product development data. If these products
have been demonstrated during development to have consistently rapid drug release
characteristics, exclusion of a particle size distribution test from the specification
may be proposed.
Particle size distribution testing may also be proposed in place of dissolution testing;
justification should be provided. The acceptance criteria should include acceptable
particle size distribution in terms of the percent of total particles in given size ranges.
The mean, upper, and / or lower particle size limits should be well defined.
Acceptance criteria should be set based on the observed range of variation, and
should take into account the dissolution profiles of the batches that showed
acceptable performance in vivo, as well as the intended use of the product. The
potential for particle growth should be investigated during product development; the
acceptance criteria should take the results of these studies into account.

Specifications: New Chemical Drug Substances and Products
15
j) Redispersibility: For oral suspensions which settle on storage (produce sediment),
acceptance criteria for redispersibility may be appropriate. Shaking may be an
appropriate procedure.
The procedure (mechanical or manual) should be indicated. Time required to achieve
resuspension by the indicated procedure should be clearly defined. Data generated
during product development may be sufficient to justify skip lot testing, or
elimination of this attribute from the specification may be proposed.
k) Rheological properties: For relatively viscous solutions or suspensions, it may be
appropriate to include rheological properties (viscosity/specific gravity) in the
specification. The test and acceptance criteria should be stated. Data generated
during product development may be sufficient to justify skip lot testing, or
elimination of this attribute from the specification may be proposed.
l) Reconstitution time: Acceptance criteria for reconstitution time should be provided
for dry powder products which require reconstitution. The choice of diluent should be
justified. Data generated during product development may be sufficient to justify skip
lot testing or elimination of this attribute from the specification may be proposed.
m) Water content: For oral products requiring reconstitution, a test and acceptance
criterion for water content should be proposed when appropriate. Loss on drying is
generally considered sufficient if the effect of absorbed moisture vs. water of
hydration has been adequately characterized during the development of the product.
In certain cases a more specific procedure (e.g., Karl Fischer titration) may be
preferable.
3.3.2.3 Parenteral Drug Products: The following tests may be applicable to
parenteral drug products.
a) Uniformity of dosage units: This term includes both the mass of the dosage form
and the content of the active substance in the dosage form; a pharmacopoeial
procedure should be used. In general, the specification should one or the other but
not both and is applicable to powders for reconstitution. When weight variation is
applied for new drug products exceeding the threshold value to allow testing
uniformity by weight variation, applicants should verify during drug development
that the homogeneity of the product is adequate.
If appropriate (see section 2.3), these tests may be performed in-process; the
acceptance criteria should be included in the specification. This test may be applied
to both single-dose and multiple-dose packages.
For powders for reconstitution, uniformity of mass testing is generally considered
acceptable.
b) pH: Acceptance criteria for pH should be provided where applicable and the
proposed range justified.
c) Sterility: All parenteral products should have a test procedure and acceptance
criterion for evaluation of sterility. Where data generated during development and
validation justify parametric release, this approach may be proposed for terminally
sterilized drug products (see section 2.6).
d) Endotoxins/Pyrogens: A test procedure and acceptance criterion for endotoxins,
using a procedure such as the limulus amoebocyte lysate test, should be included in
the specification. Pyrogenicity testing may be proposed as an alternative to
endotoxin testing where justified.
e) Particulate matter: Parenteral products should have appropriate acceptance
criteria for particulate matter. This will normally include acceptance criteria for

Specifications: New Chemical Drug Substances and Products
16
visible particulates and / or clarity of solution, as well as for sub-visible particulates
as appropriate.
f) Water content: For non-aqueous parenterals, and for parenteral products for
reconstitution, a test procedure and acceptance criterion for water content should be
proposed when appropriate. Loss on drying is generally considered sufficient for
parenteral products, if the effect of absorbed moisture vs. water of hydration has been
adequately characterized during development. In certain cases a more specific
procedure (e.g., Karl Fischer titration) may be preferred.
g) Antimicrobial preservative content: For parenteral products needing an
antimicrobial preservative, acceptance criteria for preservative content should be
established. Acceptance criteria for preservative content should be based upon the
levels of antimicrobial preservative necessary to maintain microbiological quality of
the product at all stages throughout its proposed usage and shelf life. The lowest
specified concentration of antimicrobial preservative should be demonstrated to be
effective in controlling microorganisms by using a pharmacopoeial antimicrobial
preservative effectiveness test.
Testing for antimicrobial preservative content should normally be performed at
release. Under certain circumstances, in-process testing may suffice in lieu of release
testing where permitted. When antimicrobial preservative content testing is
performed as an in-process test, the acceptance criteria should remain part of the
specification.
Antimicrobial preservative effectiveness should be demonstrated during
development, during scaleup, and throughout the shelf-life (e.g., in stability testing:
see the ICH Guideline, “Stability Testing of New Drug Substances and Products”),
although chemical testing for preservative content is the attribute normally included
in the specification.
h) Antioxidant preservative content: Release testing for antioxidant content should
normally be performed. Under certain circumstances, where justified by
developmental and stability data, shelf-life testing may be unnecessary and in-
process testing may suffice in lieu of release testing. When antioxidant content
testing is performed as an in-process test, the acceptance criteria should remain part
of the specification. If only release testing is performed, this decision should be
reinvestigated whenever either the manufacturing procedure or the container/closure
system changes.
i) Extractables: Control of extractables from container/closure systems is considered
significantly more important for parenteral products than for oral liquids. However,
where development and stability data show evidence that extractables are
consistently below the levels that are demonstrated to be acceptable and safe,
elimination of this test can normally be accepted. This should be reinvestigated if the
container/closure system or formulation changes.
Where data demonstrate the need, acceptance criteria for extractables from the
container/closure components are considered appropriate for parenteral products
packaged in non-glass systems or in glass containers with elastomeric closures. This
testing may be performed at release only, where justified by data obtained during
development. The container/closure system components (e.g., rubber stopper, etc.)
should be listed, and data collected for these components as early in the development
process as possible.
j) Functionality testing of delivery systems: Parenteral formulations packaged in pre-
filled syringes, autoinjector cartridges, or the equivalent should have test procedures

Specifications: New Chemical Drug Substances and Products
17
and acceptance criteria related to the functionality of the delivery system. These may
include control of syringeability, pressure, and seal integrity (leakage), and/or
parameters such as tip cap removal force, piston release force, piston travel force, and
power injector function force. Under certain circumstances these tests may be
performed in-process. Data generated during product development may be sufficient
to justify skip lot testing or elimination of some or all attributes from the
specification.
k) Osmolarity: When the tonicity of a product is declared in its labeling, appropriate
control of its osmolarity should be performed. Data generated during development
and validation may be sufficient to justify performance of this procedure as an in-
process control, skip lot testing, or direct calculation of this attribute.
l) Particle size distribution: Quantitative acceptance criteria and a procedure for
determination of particle size distribution may be appropriate for injectable
suspensions. Developmental data should be considered when determining the need
for either a dissolution procedure or a particle size distribution procedure.
Particle size distribution testing should be performed at release. It may be performed
as an in-process test when justified by product development data. If the product has
been demonstrated during development to have consistently rapid drug release
characteristics, exclusion of particle size controls from the specification may be
proposed.
Particle size distribution testing may also be proposed in place of dissolution testing,
when development studies demonstrate that particle size is the primary factor
influencing dissolution; justification should be provided. The acceptance criteria
should include acceptable particle size distribution in terms of the percent of total
particles in given size ranges. The mean, upper, and / or lower particle size limits
should be well defined.
Acceptance criteria should be set based on the observed range of variation, and
should take into account the dissolution profiles of the batches that showed
acceptable performance in vivo and the intended use of the product. The potential for
particle growth should be investigated during product development; the acceptance
criteria should take the results of these studies into account.
m) Redispersibility: For injectable suspensions which settle on storage (produce
sediment), acceptance criteria for redispersibility may be appropriate. Shaking may
be an appropriate procedure. The procedure (mechanical or manual) should be
indicated. Time required to achieve resuspension by the indicated procedure should
be clearly defined. Data generated during product development may be sufficient to
justify skip lot testing, or elimination of this attribute from the specification may be
proposed.
n) Reconstitution time: Acceptance criteria for reconstitution time should be provided
for all parenteral products which require reconstitution. The choice of diluent should
be justified. Data generated during product development and process validation may
be sufficient to justify skip lot testing or elimination of this attribute from the
specification for rapidly dissolving products.

Specifications: New Chemical Drug Substances and Products
18
4. GLOSSARY
(The following definitions are presented for the purpose of this Guideline)
Acceptance criteria:
Numerical limits, ranges, or other suitable measures for acceptance of the results of
analytical procedures.
Chiral:
Not superimposable with its mirror image, as applied to molecules, conformations,
and macroscopic objects, such as crystals. the term has been extended to samples of
substances whose molecules are chiral, even if the macroscopic assembly of such
molecules is racemic.
Combination product:
A drug product which contains more than one drug substance.
Degradation product:
A molecule resulting from a chemical change in the drug molecule brought about over
time and/or by the action of e.g., light, temperature, pH, water, or by reaction with an
excipient and/or the immediate container/closure system. Also called decomposition
product.
Delayed Release:
Release of a drug (or drugs) at a time other than immediately following oral
administration.
Enantiomers:
Compounds with the same molecular formula as the drug substance, which differ in
the spatial arrangement of atoms within the molecule and are nonsuperimposable
mirror images.
Extended Release:
Products which are formulated to make the drug available over an extended period
after administration.
Highly Water Soluble Drugs:
Drugs with a dose/solubility volume of less than or equal to 250 mL over a pH range
of 1.2 to 6.8. (Example: Compound A has as its lowest solubility at 37± 0.5°C, 1.0
mg/mL at pH 6.8, and is available in 100 mg, 200 mg, and 400 mg strengths. This
drug would be considered a low solubility drug as its dose/solubility volume is greater
than 250 mL (400 mg/1.0 mg/mL = 400 mL).
Immediate Release:
Allows the drug to dissolve in the gastrointestinal contents, with no intention of
delaying or prolonging the dissolution or absorption of the drug.
Impurity:
(1) Any component of the new drug substance which is not the chemical entity
defined as the new drug substance. (2) Any component of the drug product which is
not the chemical entity defined as the drug substance or an excipient in the drug
product.

Specifications: New Chemical Drug Substances and Products
19
Identified impurity:
An impurity for which a structural characterization has been achieved.
In-process tests:
Tests which may be performed during the manufacture of either the drug substance
or drug product, rather than as part of the formal battery of tests which are
conducted prior to release.
Modified Release:
Dosage forms whose drug-release characteristics of time course and/or location are
chosen to accomplish therapeutic or convenience objectives not offered by
conventional dosage forms such as a solution or an immediate release dosage form.
Modified release solid oral dosage forms include both delayed and extended release
drug products.
New drug product:
A pharmaceutical product type, for example, tablet, capsule, solution, cream, etc.,
which has not previously been registered in a region or Member State, and which
contains a drug ingredient generally, but not necessarily, in association with
excipients.
New drug substance:
The designated therapeutic moiety, which has not previously been registered in a
region or Member State (also referred to as a new molecular entity or new chemical
entity). It may be a complex, simple ester, or salt of a previously approved drug
substance.
Polymorphism:
The occurrence of different crystalline forms of the same drug substance. This may
include solvation or hydration products (also known as pseudopolymorphs) and
amorphous forms.
Quality:
The suitability of either a drug substance or drug product for its intended use. This
term includes such attributes as the identity, strength, and purity.
Racemate:
A composite (solid, liquid, gaseous, or in solution) of equimolar quantities of two
enantiomeric species. It is devoid of optical activity.
Rapidly Dissolving Products:
An immediate release solid oral drug product is considered rapidly dissolving when
not less than 80% of the label amount of the drug substance dissolves within 15
minutes in each of the following media: (1) pH 1.2, (2) pH 4.0, and (3) pH 6.8.
Reagent:
A substance, other than a starting material or solvent, which is used in the
manufacture of a new drug substance.

Specifications: New Chemical Drug Substances and Products
20
Solvent:
An inorganic or an organic liquid used as a vehicle for the preparation of solutions or
suspensions in the synthesis of a new drug substance or the manufacture of a new
drug product.
Specification:
A list of tests, references to analytical procedures, and appropriate acceptance
criteria which are numerical limits, ranges, or other criteria for the tests described. It
establishes the set of criteria to which a drug substance or drug product should
conform to be considered acceptable for its intended use. "Conformance to
specifications" means that the drug substance and / or drug product, when tested
according to the listed analytical procedures, will meet the listed acceptance criteria.
Specifications are critical quality standards that are proposed and justified by the
manufacturer and approved by regulatory authorities.
Specific test:
A test which is considered to be applicable to particular new drug substances or
particular new drug products depending on their specific properties and/or intended
use.
Specified impurity:
An identified or unidentified impurity that is selected for inclusion in the new drug
substance or new drug product specification and is individually listed and limited in
order to assure the quality of the new drug substance or new drug product.
Unidentified impurity:
An impurity which is defined solely by qualitative analytical properties, (e.g.,
chromatographic retention time).
Universal test:
A test which is considered to be potentially applicable to all new drug substances, or
all new drug products; e.g., appearance, identification, assay, and impurity tests.
5. REFERENCES
International Conference on Harmonisation; "Impurities in New Drug Substances",
1995.
International Conference on Harmonisation; "Impurities in New Drug Products",
1996.
International Conference on Harmonisation; "Stability Testing of New Drug
Substances and Products", 1994.
International Conference on Harmonisation; "Text on Validation of Analytical
Procedures", 1994.
International Conference on Harmonisation; "Validation of Analytical Procedures:
Methodology", 1996.
International Conference on Harmonisation, "Residual Solvents in Pharmaceuticals",
1996.
International Conference on Harmonisation, “Specifications: Test Procedures and
Acceptance Criteria for Biotechnological/Biological Products”, 1999
Specifications: New Chemical Drug Substances and Products
21
6. ATTACHMENTS
Decision Trees #1 through #8
Specifications: New Chemical Drug Substances and Products
22

Specifications: New Chemical Drug Substances and Products
23
Specifications: New Chemical Drug Substances and Products
24
Specifications: New Chemical Drug Substances and Products
25

Specifications: New Chemical Drug Substances and Products
26
Specifications: New Chemical Drug Substances and Products
27

Specifications: New Chemical Drug Substances and Products
28
Specifications: New Chemical Drug Substances and Products
29
Specifications: New Chemical Drug Substances and Products
30
Specifications: New Chemical Drug Substances and Products
31
